
Factores asociados al peso individual en peces demersales: un análisis con datos DATRAS en R

Cuando trabajamos con datos reales, muchas veces la parte más importante no es calcular una media o dibujar un gráfico, sino formular una pregunta de investigación que tenga sentido. En este caso, el objetivo del estudio fue analizar qué factores parecen estar más asociados con el peso individual de los ejemplares registrados en la campaña DEMERSALES 2022.
Para ello utilizamos datos biológicos reales obtenidos a través de DATRAS, la base de datos del ICES que centraliza la información de campañas oceanográficas como las realizadas por el IEO-CSIC. A partir de estos registros, el trabajo se planteó como una aplicación práctica de herramientas clásicas de Estadística Matemática sobre un conjunto de datos reales: análisis exploratorio, intervalos de confianza, contrastes de hipótesis y modelos lineales sencillos.
La idea inicial era comparar varias especies de interés pesquero, pero tras la depuración de los datos la muestra analítica final quedó formada principalmente por Merluccius merluccius (merluza) y Trachurus trachurus (jurel). A partir de ahí, el estudio dejó de centrarse solo en una comparación descriptiva entre especies y pasó a una pregunta más interesante desde el punto de vista estadístico:
¿Qué factores explican mejor el peso individual de los ejemplares analizados: la especie, la longitud o la edad?
Esta reformulación da bastante más valor al análisis. En lugar de quedarse en una comparación obvia entre especies con tamaños distintos, el trabajo trata de evaluar el papel de tres posibles factores explicativos:
- la especie del ejemplar,
- su longitud,
- y su edad.
A lo largo del estudio construimos una muestra biométrica válida a partir de la API de DATRAS, describimos las distribuciones de peso, longitud y edad, analizamos si la especie introduce diferencias estadísticamente significativas en el peso medio y, finalmente, comparamos varios modelos sencillos para valorar qué variable parece aportar mayor capacidad explicativa sobre el peso individual.
1) Extracción de datos
Una vez definida la pregunta de investigación, lo primero que necesitamos es una
muestra biométrica válida. En nuestro caso no partimos de un CSV descargado manualmente,
sino que accedemos directamente a los datos a través de la API de DATRAS usando el paquete
icesDatras en R.
Esto tiene una ventaja importante: el flujo de trabajo es reproducible. Si alguien quisiera repetir exactamente el análisis, podría hacerlo ejecutando el mismo código y consultando la misma fuente oficial.
1.1) Cargar paquetes y conectarnos a DATRAS
El primer paso consiste en cargar los paquetes que vamos a utilizar. Aquí icesDatras se encarga de la
consulta a DATRAS, mientras que dplyr y ggplot2 nos servirán más adelante para manipular y
visualizar los datos.
# install.packages("icesDatras")
library(icesDatras)
library(dplyr)
library(ggplot2)
library(knitr)
A continuación descargamos los registros biológicos de la campaña SP-NORTH para 2022. En lugar de hacer una
única consulta agregada, descargamos los datos por trimestres y luego los unimos en una sola tabla. Esta estrategia nos
permite controlar mejor la extracción y comprobar qué información devuelve cada consulta.
# Descargamos los datos de cada trimestre de 2022
q1 <- getDATRAS(record = "CA", survey = "SP-NORTH", years = 2022, quarters = 1)
q2 <- suppressMessages(suppressWarnings(getDATRAS(record = "CA", survey = "SP-NORTH", years = 2022, quarters = 2)))
q3 <- suppressMessages(suppressWarnings(getDATRAS(record = "CA", survey = "SP-NORTH", years = 2022, quarters = 3)))
q4 <- suppressMessages(suppressWarnings(getDATRAS(record = "CA", survey = "SP-NORTH", years = 2022, quarters = 4)))
# Unimos todos los trimestres en una sola tabla
datos_raw <- rbind(q1, q2, q3, q4)1.2) Qué tipo de datos estamos descargando
Aquí estamos consultando el registro "CA", que corresponde a datos biológicos individuales
(Catch at Age / Biology). Eso significa que cada fila no representa una captura agregada ni una media por lance,
sino un ejemplar concreto para el que se han registrado variables como la especie, la longitud, el peso o,
en algunos casos, la edad.
Por eso, nuestra unidad de análisis es cada pez observado individualmente. Este detalle es importante, porque condiciona todo lo que haremos después: histogramas, intervalos de confianza, contrastes de hipótesis y modelos lineales se aplicarán sobre observaciones individuales, no sobre promedios agregados.
1.3) Primera inspección de la tabla
Antes de limpiar nada, conviene entender qué hemos descargado realmente. Por eso imprimimos el tamaño de la tabla y los nombres exactos de las columnas disponibles.
# Imprimir dimensiones
cat("TOTAL DE DATOS DESCARGADOS:\n")
cat("Filas:", nrow(datos_raw), "- Columnas:", ncol(datos_raw), "\n\n")
# Imprimir todos los nombres de las columnas
cat("NOMBRES EXACTOS DE LAS COLUMNAS:\n")
cat(names(datos_raw))Esta comprobación previa es más importante de lo que parece. En datasets reales no siempre todas las variables vienen bien documentadas ni disponibles con el mismo formato. Por eso, antes de construir la muestra analítica, verificamos qué campos están realmente presentes y cómo aparecen nombrados en la extracción.
1.4) Calidad del dato y variables biométricas
Una vez descargada la tabla bruta, hacemos una revisión mínima de la calidad del dato. Nos interesa especialmente comprobar tres cosas:
- cuántos registros biológicos se han descargado en total,
- cuántas especies distintas aparecen,
- y cuántos registros tienen problemas en variables clave como el peso individual.
# Volumen total de la campaña
cat("Total de registros biológicos procesados:", nrow(datos_raw), "\n")
# Biodiversidad
num_especies <- length(unique(datos_raw$Valid_Aphia))
cat("Número de especies distintas (AphiaIDs) capturadas:", num_especies, "\n")
# Calidad del dato: peso individual
pesos_invalidos <- sum(is.na(datos_raw$IndWgt) | datos_raw$IndWgt <= 0)
porcentaje_invalidos <- round((pesos_invalidos / nrow(datos_raw)) * 100, 2)
cat("Registros sin un peso individual válido (NAs o negativos):", pesos_invalidos,
"(", porcentaje_invalidos, "% del total)\n")
Aquí hay un detalle importante: en bases de datos reales es habitual que algunos valores no estén medidos o aparezcan
codificados de forma especial. En nuestro caso, el peso individual IndWgt puede venir ausente o con valores no
válidos, por lo que filtrar esta variable es imprescindible antes de cualquier análisis biométrico serio.
1.5) Identificar las especies presentes
Después de revisar la calidad general, inspeccionamos la columna Valid_Aphia, que identifica taxonómicamente
cada ejemplar. Esto nos permite saber qué especies están realmente presentes en la extracción y decidir cuáles tienen sentido
para el estudio.
table(datos_raw$Valid_Aphia)A partir de esta tabla construimos un pequeño diccionario taxonómico y seleccionamos inicialmente tres especies de interés:
- Merluccius merluccius (merluza),
- Micromesistius poutassou (bacaladilla),
- Trachurus trachurus (jurel).
Esta selección es todavía provisional. En este punto no sabemos aún si las tres especies tendrán suficientes observaciones válidas una vez apliquemos los filtros biométricos. Esa decisión final la marcará la limpieza de datos.
1.6) Limpieza de la muestra y construcción de la tabla analítica
Una vez elegidas las especies candidatas, aplicamos los filtros mínimos necesarios para construir una muestra con sentido estadístico. En concreto, exigimos:
- que el ejemplar pertenezca a una de las especies seleccionadas,
- que el peso individual sea válido y positivo,
- y que exista información de longitud.
# Usamos los códigos reales identificados en nuestra exploración
especies_ids <- c(126484, 126439, 126822)
# Aplicamos los filtros mínimos de limpieza
datos_limpios <- subset(
datos_raw,
Valid_Aphia %in% especies_ids &
!is.na(IndWgt) & IndWgt > 0 &
!is.na(LngtClass)
)Este paso es fundamental porque aquí es donde pasamos de una tabla biológica bruta a una muestra realmente utilizable para el análisis. No estamos “maquillando” los datos, sino eliminando observaciones que no permitirían estudiar correctamente el peso individual.
1.7) Renombrar variables y construir una tabla más cómoda de trabajar
La tabla original de DATRAS contiene nombres de variables muy útiles desde el punto de vista técnico, pero poco cómodos para
el análisis y la visualización. Por eso construimos una nueva tabla, datos_ieo, con nombres más claros y con las
unidades ya adaptadas.
# Mapeamos los códigos numéricos a texto para que los gráficos sean legibles
datos_limpios$Especie <- ifelse(
datos_limpios$Valid_Aphia == 126484, "Merluccius merluccius (Merluza)",
ifelse(datos_limpios$Valid_Aphia == 126439, "Micromesistius poutassou (Bacaladilla)",
"Trachurus trachurus (Jurel)")
)
# Creación de la tabla de análisis final
datos_ieo <- data.frame(
ID_Pez = seq_len(nrow(datos_limpios)),
Especie = datos_limpios$Especie,
Longitud_cm = datos_limpios$LngtClass / 10,
Peso_g = datos_limpios$IndWgt,
Edad = suppressWarnings(as.numeric(as.character(datos_limpios$Age)))
)
Aquí hay un detalle que conviene aclarar: la variable ID_Pez no es un identificador biológico real,
ni una especie de “DNI del pez”. Es simplemente un índice artificial que creamos nosotros para numerar las filas
de la muestra y poder referirnos a cada observación individual de forma cómoda durante el análisis.
También hay dos transformaciones importantes:
LngtClass / 10: convertimos la longitud a centímetros para trabajar con una unidad más interpretable.Age→Edad: intentamos convertir la edad a formato numérico, asumiendo que no todos los registros vendrán limpios.
1.8) Qué pasó finalmente con las especies seleccionadas
Una vez construida la tabla analítica, comprobamos cuántos registros válidos han sobrevivido al filtrado y cómo se reparten entre especies.
cat("Muestra final válida para el análisis biométrico:", nrow(datos_ieo), "peces.\n")
cat("Registros con edad válida:", sum(!is.na(datos_ieo$Edad) & datos_ieo$Edad >= 0), "\n")# Ordenamos las especies para que salgan siempre igual en tablas y gráficos
datos_ieo$Especie <- factor(
datos_ieo$Especie,
levels = c(
"Merluccius merluccius (Merluza)",
"Micromesistius poutassou (Bacaladilla)",
"Trachurus trachurus (Jurel)"
)
)
table(datos_ieo$Especie)Aquí aparece uno de los hallazgos metodológicos importantes del trabajo: aunque inicialmente seleccionamos tres especies, la bacaladilla no aportó observaciones válidas en la muestra final. En consecuencia, el análisis principal termina centrándose en merluza y jurel.
Además, la edad no quedó disponible de la misma forma para todas las especies, lo que después condicionará la parte inferencial. Esto es algo muy habitual en datos reales: la pregunta de investigación inicial puede ser ambiciosa, pero la calidad y disponibilidad efectiva de las variables acaba marcando qué comparaciones pueden hacerse de forma rigurosa y cuáles no.
Con esta limpieza ya tenemos lista la base del estudio: una tabla biométrica consistente, construida a partir de datos reales, sobre la que sí tiene sentido empezar el análisis exploratorio.
2) Análisis exploratorio de datos
Una vez construida la muestra analítica, el siguiente paso consiste en estudiar cómo se distribuyen las variables principales del trabajo: especie, peso individual, longitud y edad. El objetivo aquí no es todavía “demostrar” nada de forma inferencial, sino entender bien la estructura de la muestra, detectar asimetrías, valores extremos y relaciones entre variables, y comprobar si la pregunta de investigación tiene sentido con los datos que realmente tenemos.
En este caso, el análisis exploratorio resulta especialmente importante por dos motivos. Primero, porque trabajamos con datos reales y no con un dataset limpio de manual, así que necesitamos ver qué información aportan de verdad las variables disponibles. Segundo, porque la propia pregunta del estudio —qué factor parece explicar mejor el peso individual— exige observar no solo diferencias entre especies, sino también la relación del peso con la longitud y con la edad.
2.1) Composición de la muestra
Lo primero que hicimos fue comprobar cómo quedaba repartida la muestra final por especie. Para ello calculamos las frecuencias absolutas y relativas de cada grupo.
tabla_especies <- datos_ieo %>%
count(Especie, name = "Frecuencia") %>%
mutate(Porcentaje = round(100 * Frecuencia / sum(Frecuencia), 2))
print(tabla_especies)
ggplot(tabla_especies, aes(x = Especie, y = Frecuencia)) +
geom_col(fill = "steelblue") +
labs(
title = "Número de ejemplares por especie",
x = "Especie",
y = "Frecuencia"
) +
theme_minimal()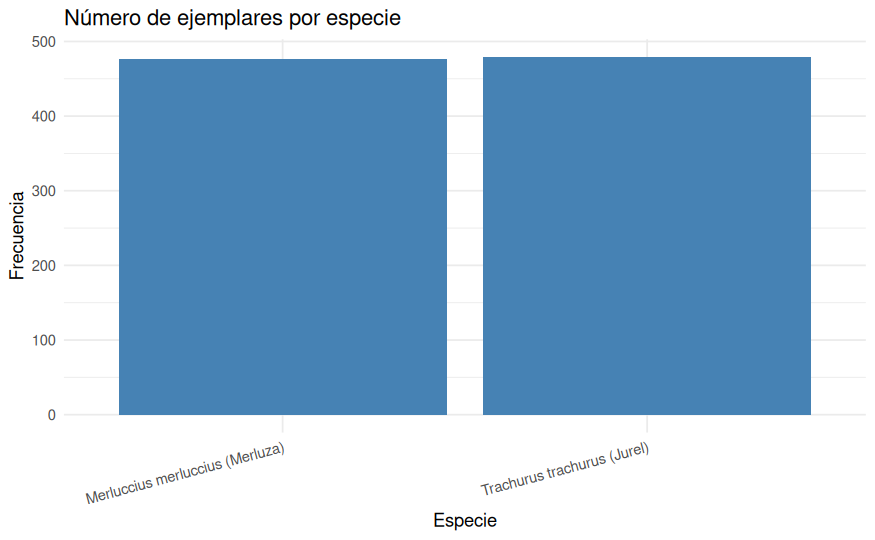
El resultado muestra una muestra prácticamente equilibrada: 477 ejemplares de merluza y 479 de jurel. Este reparto es conveniente para el estudio porque evita que una especie domine claramente en tamaño muestral y hace más razonables las comparaciones posteriores.
2.2) Resumen descriptivo global y por especie
Después de comprobar la composición de la muestra, calculamos un resumen descriptivo básico para ver la posición central y la dispersión de las variables principales.
resumen_global <- datos_ieo %>%
summarise(
n = n(),
media_peso = mean(Peso_g),
mediana_peso = median(Peso_g),
sd_peso = sd(Peso_g),
media_longitud = mean(Longitud_cm),
mediana_longitud = median(Longitud_cm),
sd_longitud = sd(Longitud_cm),
media_edad = mean(Edad, na.rm = TRUE),
mediana_edad = median(Edad, na.rm = TRUE),
sd_edad = sd(Edad, na.rm = TRUE)
) %>%
mutate(across(where(is.numeric), ~ round(.x, 2)))
print(resumen_global)
resumen_especies <- datos_ieo %>%
group_by(Especie) %>%
summarise(
n = n(),
media_peso = mean(Peso_g),
mediana_peso = median(Peso_g),
sd_peso = sd(Peso_g),
cv_peso = 100 * sd(Peso_g) / mean(Peso_g),
media_longitud = mean(Longitud_cm),
mediana_longitud = median(Longitud_cm),
sd_longitud = sd(Longitud_cm),
media_edad = mean(Edad, na.rm = TRUE),
mediana_edad = median(Edad, na.rm = TRUE),
sd_edad = sd(Edad, na.rm = TRUE)
) %>%
mutate(across(where(is.numeric), ~ round(.x, 2)))
print(resumen_especies)A nivel global, la muestra final quedó formada por 956 ejemplares. El peso medio conjunto fue de 310.29 gramos, aunque la mediana se situó en 118 gramos, lo que ya nos adelanta una distribución muy asimétrica a la derecha. La desviación típica del peso alcanzó 531.94 gramos, un valor muy elevado que confirma una gran dispersión. En longitud, la media global fue de 27.62 cm y la mediana de 25.25 cm.
Cuando separamos por especie, las diferencias biométricas son claras. La merluza presenta un peso medio cercano a 525 gramos, frente a los 96.6 gramos del jurel, y una longitud media aproximada de 36.6 cm, claramente superior a la del jurel. Además, la merluza muestra una dispersión bastante mayor, lo que ya sugiere que la especie podría estar asociada al peso, aunque todavía no podamos decir si es el factor más informativo.
2.3) Distribución del peso individual
Para estudiar la forma de la distribución del peso individual comenzamos con un histograma global y, después, repetimos el análisis separando por especie.
ggplot(datos_ieo, aes(x = Peso_g)) +
geom_histogram(bins = 30, fill = "darkcyan", color = "black") +
labs(
title = "Distribución global del peso individual",
x = "Peso individual (g)",
y = "Frecuencia"
) +
theme_minimal()
ggplot(datos_ieo, aes(x = Peso_g)) +
geom_histogram(bins = 25, fill = "darkorange", color = "black") +
facet_wrap(~ Especie, scales = "free") +
labs(
title = "Distribución del peso individual por especie",
x = "Peso individual (g)",
y = "Frecuencia"
) +
theme_minimal()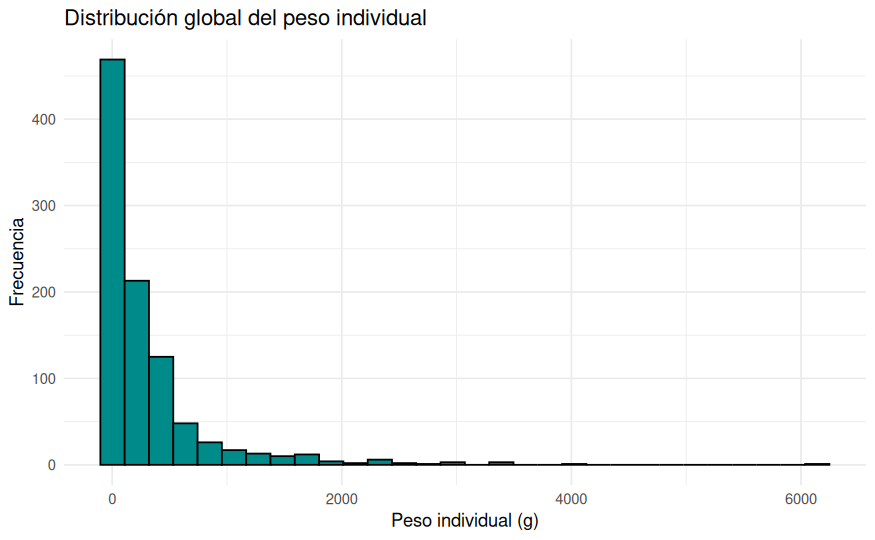
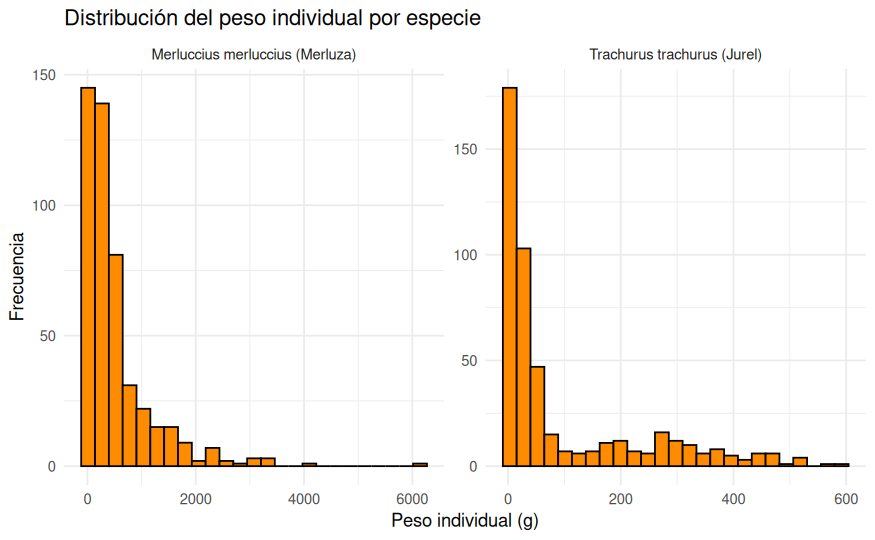
El histograma global muestra una asimetría muy marcada a la derecha: la mayor parte de los ejemplares se concentra en pesos bajos, mientras que existe un número reducido de individuos con pesos muy elevados. Cuando separamos por especie, esta estructura se entiende mejor. Tanto merluza como jurel presentan distribuciones asimétricas, pero la merluza exhibe una cola derecha mucho más larga y una variabilidad mucho mayor. Esto encaja con lo ya visto en las tablas descriptivas.
2.4) Distribución de la longitud
Como la longitud es una de las variables candidatas a explicar el peso, su distribución merece una inspección específica.
ggplot(datos_ieo, aes(x = Longitud_cm)) +
geom_histogram(bins = 25, fill = "mediumpurple", color = "black") +
facet_wrap(~ Especie, scales = "free") +
labs(
title = "Distribución de la longitud por especie",
x = "Longitud (cm)",
y = "Frecuencia"
) +
theme_minimal()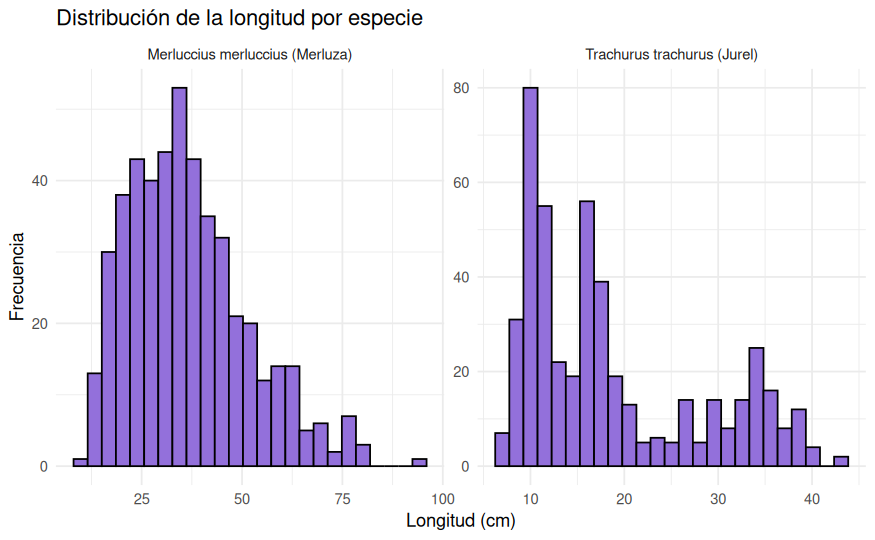
La longitud también muestra diferencias claras entre especies. La merluza presenta longitudes superiores y una dispersión más amplia, mientras que el jurel queda concentrado en tamaños más reducidos. Desde un punto de vista biométrico, esto ya hace razonable pensar que parte de la diferencia de peso puede estar muy ligada al tamaño del ejemplar y no solo a la especie en sí.
2.5) Distribución de la edad
La edad introduce una dimensión biológica adicional al estudio, pero aquí aparece una limitación importante de los datos: tras el filtrado, solo disponemos de registros de edad válidos para el jurel.
datos_edad <- datos_ieo %>%
filter(!is.na(Edad), Edad >= 0)
print(datos_edad %>% count(Especie))
ggplot(datos_edad, aes(x = Edad)) +
geom_histogram(binwidth = 1, fill = "goldenrod", color = "black") +
facet_wrap(~ Especie, scales = "free") +
labs(
title = "Distribución de la edad por especie",
x = "Edad",
y = "Frecuencia"
) +
theme_minimal()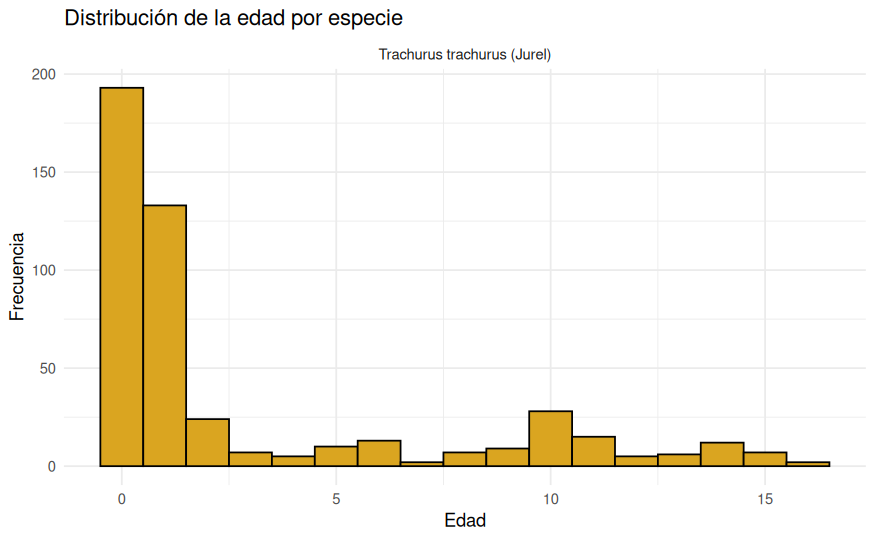
En la práctica, esto significa que la edad solo puede analizarse sobre una submuestra de 478 jureles. Por tanto, podremos estudiar si la edad está asociada con el peso dentro de esa submuestra, pero no comparar su efecto entre especies en igualdad de condiciones.
2.6) Comparación visual entre especies
Para complementar los histogramas utilizamos diagramas de caja, que resumen muy bien la posición central, la dispersión y la presencia de valores atípicos.
ggplot(datos_ieo, aes(x = Especie, y = Peso_g)) +
geom_boxplot(fill = "tomato", alpha = 0.8, outlier.alpha = 0.6) +
labs(
title = "Comparación del peso individual entre especies",
x = "Especie",
y = "Peso individual (g)"
) +
theme_minimal()
ggplot(datos_ieo, aes(x = Especie, y = Longitud_cm)) +
geom_boxplot(fill = "seagreen", alpha = 0.8, outlier.alpha = 0.6) +
labs(
title = "Comparación de la longitud entre especies",
x = "Especie",
y = "Longitud (cm)"
) +
theme_minimal()
ggplot(datos_edad, aes(x = Especie, y = Edad)) +
geom_boxplot(fill = "khaki4", alpha = 0.8, outlier.alpha = 0.6) +
labs(
title = "Comparación de la edad entre especies",
x = "Especie",
y = "Edad"
) +
theme_minimal()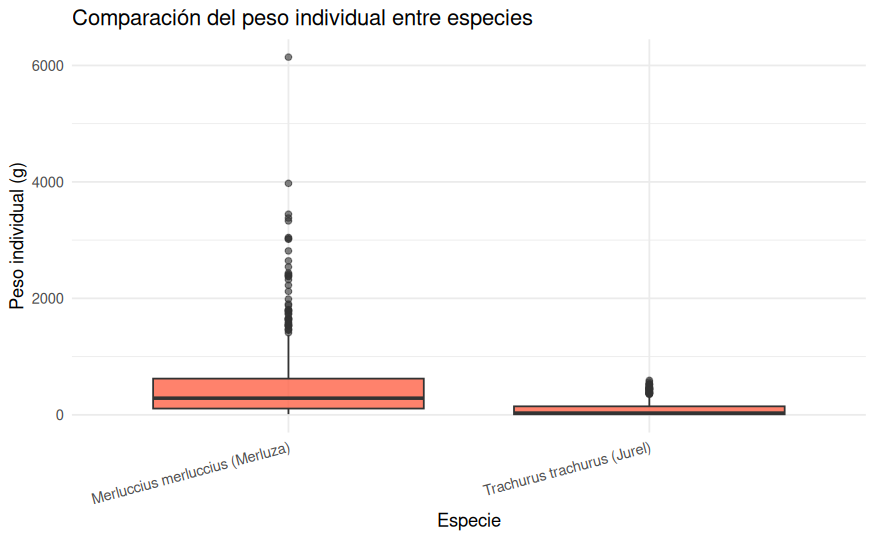
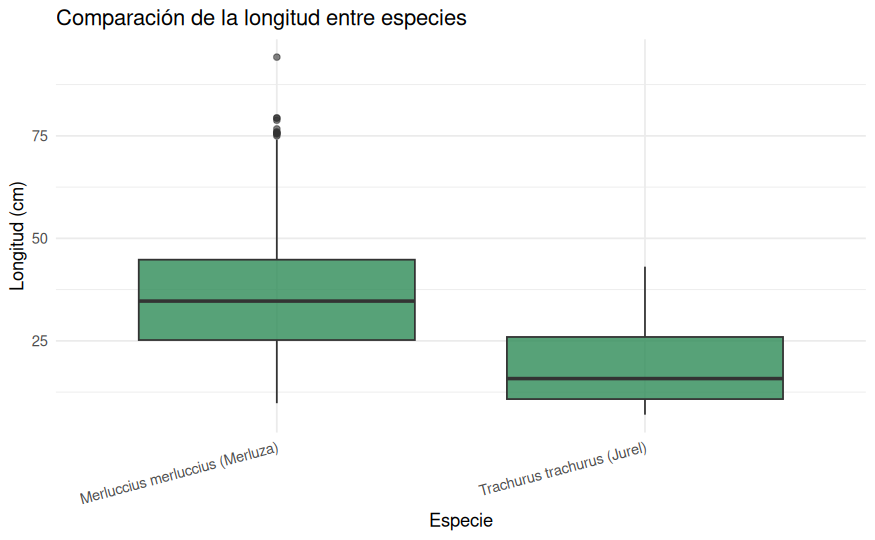
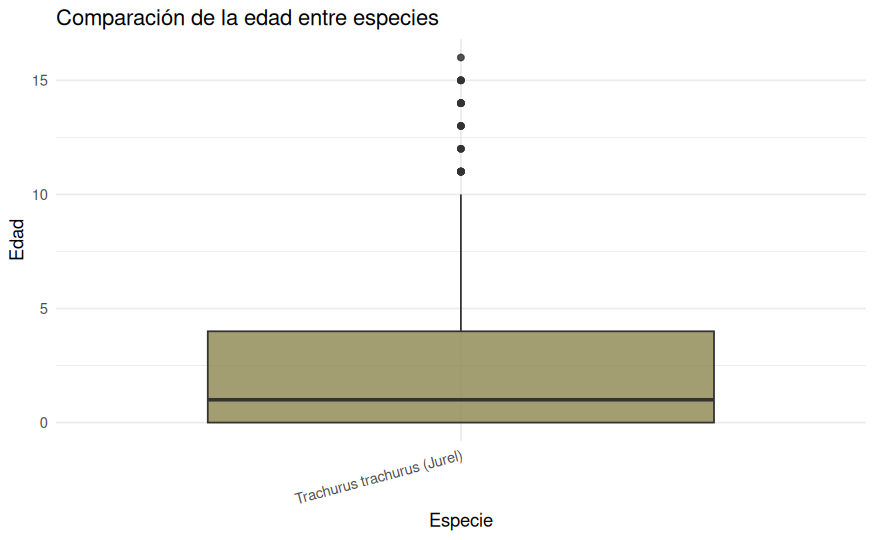
Los boxplots refuerzan lo observado hasta ahora. La merluza presenta medianas de peso y longitud claramente superiores a las del jurel, además de una dispersión mucho mayor y más valores extremos. En cambio, la edad solo puede visualizarse para el jurel, precisamente porque no quedaron edades válidas para la merluza en la muestra final.
2.7) Relación entre longitud y peso
Una vez vistas las distribuciones marginales, el siguiente paso consiste en estudiar la relación entre las variables. Empezamos por la longitud, que desde el principio parecía la candidata más fuerte a estar asociada con el peso.
ggplot(datos_ieo, aes(x = Longitud_cm, y = Peso_g)) +
geom_point(alpha = 0.6, size = 1.8) +
geom_smooth(method = "lm", se = FALSE, linewidth = 0.8) +
facet_wrap(~ Especie, scales = "free") +
labs(
title = "Relación entre longitud y peso por especie",
x = "Longitud (cm)",
y = "Peso individual (g)"
) +
theme_minimal()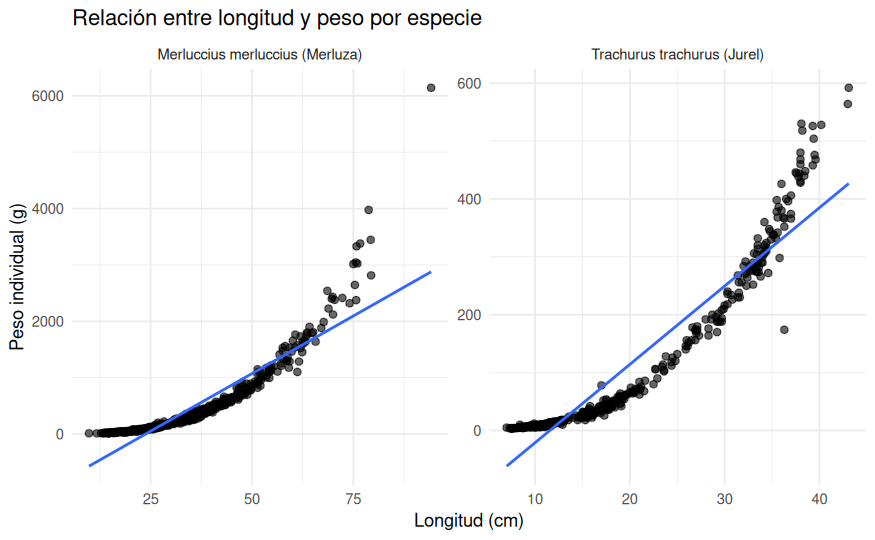
El diagrama de dispersión muestra una relación positiva muy clara entre longitud y peso en ambas especies: en general, a mayor longitud corresponde un mayor peso individual. Visualmente, este es probablemente el patrón más fuerte de todo el análisis exploratorio y ya nos adelanta que la longitud podría ser el factor más informativo entre los considerados.
2.8) Relación entre edad y peso
Repetimos la misma idea para la edad, aunque en este caso debemos recordar que solo trabajamos con la submuestra de jureles con edad válida.
ggplot(datos_edad, aes(x = Edad, y = Peso_g)) +
geom_point(alpha = 0.6, size = 1.8) +
geom_smooth(method = "lm", se = FALSE, linewidth = 0.8) +
facet_wrap(~ Especie, scales = "free") +
labs(
title = "Relación entre edad y peso por especie",
x = "Edad",
y = "Peso individual (g)"
) +
theme_minimal()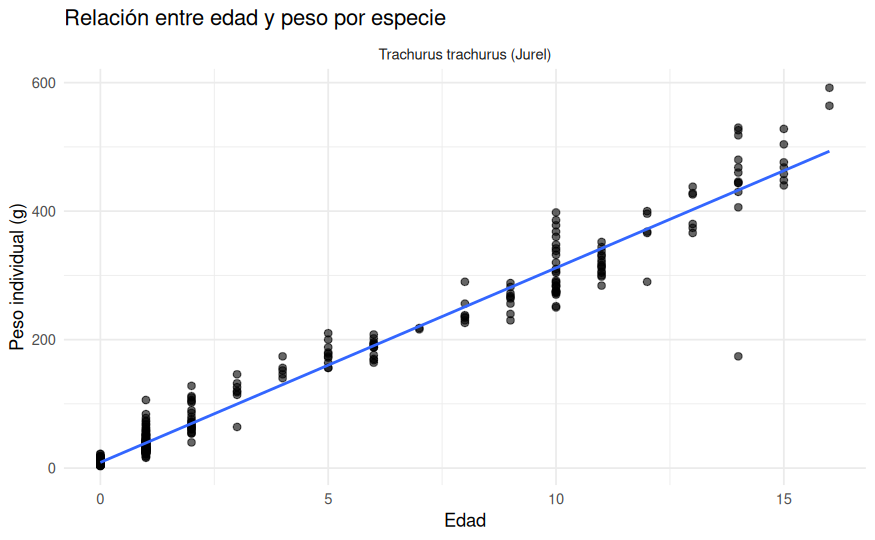
En esta submuestra también se aprecia una relación creciente: los individuos de mayor edad tienden a presentar mayor peso. Sin embargo, el análisis de la edad tiene menos fuerza que el de la longitud porque no puede compararse entre especies y porque depende de una submuestra mucho más limitada.
2.9) Correlaciones biométricas
Para cuantificar con más precisión lo que sugieren los gráficos anteriores, calculamos coeficientes de correlación de Pearson y Spearman.
correlaciones_longitud <- datos_ieo %>%
group_by(Especie) %>%
summarise(
Correlacion_Pearson = cor(Longitud_cm, Peso_g, method = "pearson"),
Correlacion_Spearman = cor(Longitud_cm, Peso_g, method = "spearman")
) %>%
mutate(across(where(is.numeric), ~ round(.x, 4)))
print(correlaciones_longitud)
correlaciones_edad <- datos_edad %>%
group_by(Especie) %>%
summarise(
Correlacion_Pearson = cor(Edad, Peso_g, method = "pearson"),
Correlacion_Spearman = cor(Edad, Peso_g, method = "spearman")
) %>%
mutate(across(where(is.numeric), ~ round(.x, 4)))
print(correlaciones_edad)Los resultados confirman de forma numérica lo observado visualmente. La longitud muestra una asociación muy fuerte con el peso en ambas especies: la correlación de Pearson es 0.904 en la merluza y 0.951 en el jurel, mientras que la de Spearman alcanza 0.996 y 0.988, respectivamente. Por su parte, en la submuestra con edad válida la relación edad-peso en el jurel también es intensa, con una correlación de Pearson de 0.985 y una de Spearman de 0.953. Aun así, esta última evidencia debe interpretarse con cautela por la limitación ya comentada.
2.10) Asimetría y transformación logarítmica del peso
Antes de pasar a la parte inferencial, también comprobamos hasta qué punto el peso individual se aleja de una distribución aproximadamente normal y si una transformación logarítmica mejora esa situación.
ggplot(datos_ieo, aes(sample = Peso_g)) +
stat_qq(size = 1, alpha = 0.6) +
stat_qq_line(color = "red", linewidth = 0.7) +
facet_wrap(~ Especie, scales = "free") +
labs(
title = "Gráficos Q-Q del peso individual por especie",
x = "Cuantiles teóricos",
y = "Cuantiles muestrales"
) +
theme_minimal()
datos_ieo <- datos_ieo %>%
mutate(log_Peso_g = log(Peso_g))
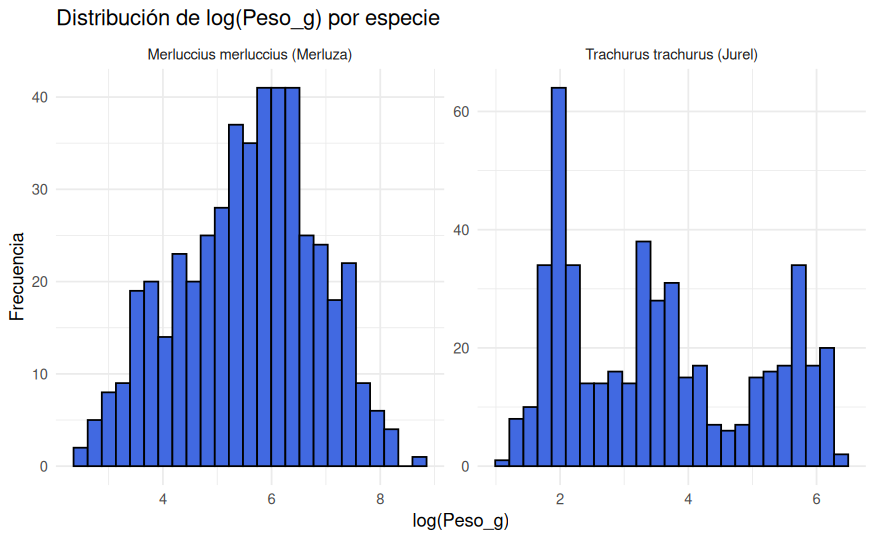
ggplot(datos_ieo, aes(x = log_Peso_g)) +
geom_histogram(bins = 25, fill = "royalblue", color = "black") +
facet_wrap(~ Especie, scales = "free") +
labs(
title = "Distribución de log(Peso_g) por especie",
x = "log(Peso_g)",
y = "Frecuencia"
) +
theme_minimal()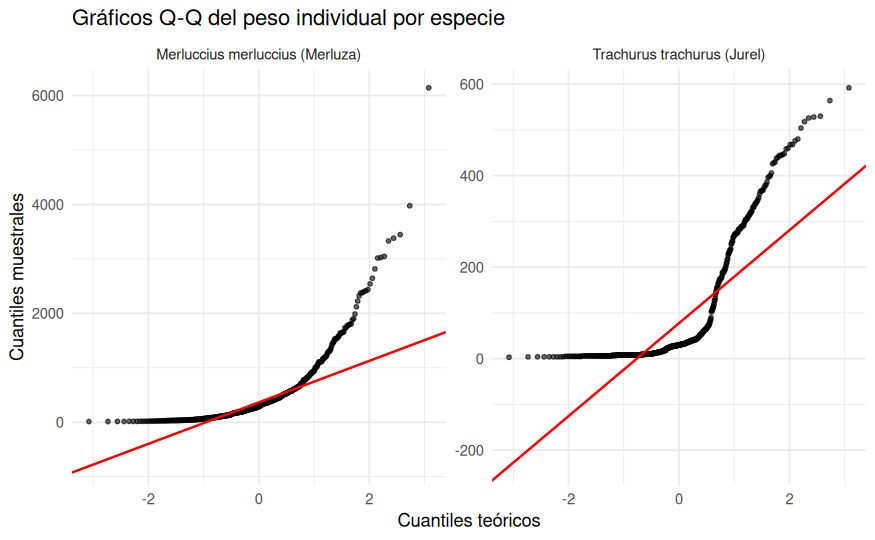
Los gráficos Q-Q muestran desviaciones claras respecto a la normalidad, especialmente por la cola derecha del peso.
Esto era esperable a la vista de los histogramas. La transformación log(Peso_g) reduce esa asimetría y deja
una variable más estable para trabajar en los modelos posteriores.
En conjunto, el análisis exploratorio deja una idea bastante clara. La especie parece asociarse con el peso porque merluza y jurel presentan biometrías muy distintas, pero la longitud emerge como la variable más estrechamente conectada con el peso individual. La edad también muestra una relación fuerte dentro del jurel, aunque con una evidencia más limitada por la estructura de los datos disponibles. Con este contexto, ya sí tiene sentido pasar a la parte inferencial del estudio.
3) Inferencia estadística
Una vez entendido el comportamiento general de la muestra, el siguiente paso consiste en pasar de la descripción a la inferencia estadística. Hasta ahora hemos visto patrones claros en los gráficos y en las tablas, pero todavía no hemos cuantificado formalmente la incertidumbre asociada a esos resultados ni hemos evaluado hasta qué punto las diferencias observadas pueden atribuirse al azar muestral.
En esta parte del estudio reinterpretemos los datos desde una perspectiva inferencial. Nuestro objetivo ya no es solo describir la muestra, sino estudiar qué variables parecen desempeñar un papel más relevante en la explicación del peso individual. Para ello combinamos tres herramientas principales:
- estimación puntual, para resumir el comportamiento medio de cada especie,
- intervalos de confianza, para cuantificar la incertidumbre de las estimaciones,
- y contrastes de hipótesis y modelos lineales sencillos, para valorar el papel de la especie, la longitud y la edad.
3.1) Efecto de la especie sobre el peso individual
Aunque la pregunta general del trabajo es más amplia, la especie sigue siendo uno de los factores candidatos a explicar diferencias de peso. Por eso empezamos analizando si merluza y jurel presentan diferencias estadísticamente significativas en su peso medio.
3.1.1) Estimación puntual por especie
El primer paso consiste en calcular estimadores puntuales básicos para cada grupo. En nuestro caso, las medias muestrales ya muestran una separación muy clara: la merluza presenta un peso medio de 524.83 gramos, mientras que el jurel se sitúa en 96.65 gramos. La diferencia observada entre ambas medias es, por tanto, de 428.18 gramos.
estimadores_puntuales <- datos_ieo %>%
group_by(Especie) %>%
summarise(
n = n(),
media_peso = round(mean(Peso_g), 2),
varianza_peso = round(var(Peso_g), 2),
sd_peso = round(sd(Peso_g), 2),
media_longitud = round(mean(Longitud_cm), 2),
varianza_longitud = round(var(Longitud_cm), 2),
sd_longitud = round(sd(Longitud_cm), 2)
)
print(estimadores_puntuales)Esta tabla permite dar un primer paso hacia la inferencia: ya no estamos usando la media solo como descripción de la muestra, sino como estimación puntual de la media poblacional del peso en cada especie.
3.1.2) Intervalos de confianza para la media del peso
Para cuantificar la incertidumbre de estas estimaciones construimos intervalos de confianza al 95% para la media del peso individual de cada especie. Como la varianza poblacional es desconocida, utilizamos la distribución t de Student.
# Merluza
y <- datos_ieo$Peso_g[datos_ieo$Especie == "Merluccius merluccius (Merluza)"]
m <- length(y)
media_muestral <- mean(y)
desviacion_tipica <- sd(y)
alpha <- 0.05
gl <- m - 1
t_critico <- qt(1 - alpha/2, df = gl)
se_media <- desviacion_tipica / sqrt(m)
margen_error <- t_critico * se_media
ic_inferior <- media_muestral - margen_error
ic_superior <- media_muestral + margen_error
cat("IC95% para la media del peso de la merluza = (",
round(ic_inferior, 4), ",", round(ic_superior, 4), ")\n")
# Jurel
y <- datos_ieo$Peso_g[datos_ieo$Especie == "Trachurus trachurus (Jurel)"]
m <- length(y)
media_muestral <- mean(y)
desviacion_tipica <- sd(y)
gl <- m - 1
t_critico <- qt(1 - alpha/2, df = gl)
se_media <- desviacion_tipica / sqrt(m)
margen_error <- t_critico * se_media
ic_inferior <- media_muestral - margen_error
ic_superior <- media_muestral + margen_error
cat("IC95% para la media del peso del jurel = (",
round(ic_inferior, 4), ",", round(ic_superior, 4), ")\n")Los resultados son bastante claros. El intervalo de confianza del 95% para la media del peso de la merluza es (463.97, 585.68) gramos, mientras que para el jurel es (84.59, 108.71) gramos. Además de mostrar que ambas medias están muy separadas, estos intervalos permiten cuantificar de forma explícita la incertidumbre de las estimaciones.
3.1.3) Intervalo de confianza para la diferencia de medias
Más allá de estimar cada especie por separado, también nos interesa cuantificar directamente la diferencia entre sus medias poblacionales. Como las varianzas no parecen iguales, utilizamos el procedimiento de Welch.
peso_merluza <- datos_ieo$Peso_g[datos_ieo$Especie == "Merluccius merluccius (Merluza)"]
peso_jurel <- datos_ieo$Peso_g[datos_ieo$Especie == "Trachurus trachurus (Jurel)"]
alpha <- 0.05
n1 <- length(peso_merluza)
n2 <- length(peso_jurel)
xbar1 <- mean(peso_merluza)
xbar2 <- mean(peso_jurel)
s1_2 <- var(peso_merluza)
s2_2 <- var(peso_jurel)
dif_medias <- xbar1 - xbar2
se_dif <- sqrt((s1_2 / n1) + (s2_2 / n2))
gl_welch <- ((s1_2 / n1 + s2_2 / n2)^2) /
(((s1_2 / n1)^2) / (n1 - 1) + ((s2_2 / n2)^2) / (n2 - 1))
t_critico <- qt(1 - alpha/2, df = gl_welch)
margen_error <- t_critico * se_dif
ic_inf <- dif_medias - margen_error
ic_sup <- dif_medias + margen_error
cat("IC95% para la diferencia de medias (Merluza - Jurel) = (",
round(ic_inf, 4), ",", round(ic_sup, 4), ")\n")El intervalo de confianza al 95% para la diferencia de medias del peso individual entre merluza y jurel es (366.15, 490.21) gramos. Como el intervalo no contiene el valor 0 y todos sus valores son positivos, la evidencia apunta de forma clara a que la merluza presenta un peso medio superior al del jurel. En términos prácticos, la diferencia media estimada entre ambas especies se sitúa entre 366 y 490 gramos.
3.1.4) Contraste de hipótesis para comparar medias entre especies
Aunque el análisis exploratorio ya sugería diferencias claras, el contraste de hipótesis permite formalizar esa evidencia dentro del marco clásico de la inferencia estadística. Dado que observamos dispersiones distintas entre especies, utilizamos de nuevo el test t de Welch.
resultado_welch <- t.test(
peso_merluza,
peso_jurel,
alternative = "two.sided",
var.equal = FALSE,
conf.level = 0.95
)
t_estadistico <- resultado_welch$statistic
gl_welch <- resultado_welch$parameter
p_valor <- resultado_welch$p.value
ic_contraste <- resultado_welch$conf.int
dif_medias <- mean(peso_merluza) - mean(peso_jurel)
cat("Diferencia de medias (Merluza - Jurel) =", round(dif_medias, 4), "\n")
cat("Estadístico t =", round(t_estadistico, 4), "\n")
cat("Grados de libertad (Welch) =", round(gl_welch, 4), "\n")
cat("p-valor =", round(p_valor, 6), "\n")
cat("IC95% para la diferencia de medias = (",
round(ic_contraste[1], 4), ",", round(ic_contraste[2], 4), ")\n")El resultado del test es concluyente: la diferencia de medias estimada es de 428.18 gramos, el estadístico observado vale 13.5613, los grados de libertad aproximados son 513.34 y el p-valor es prácticamente nulo. En consecuencia, rechazamos la hipótesis nula de igualdad de medias y concluimos que existe una diferencia estadísticamente significativa entre el peso medio de la merluza y el del jurel.
Hasta aquí, por tanto, la especie sí parece desempeñar un papel importante. Sin embargo, esto no responde todavía a la pregunta principal del estudio: que la especie sea relevante no implica necesariamente que sea el factor que mejor explique el peso individual.
3.2) Asociación entre longitud y peso
La longitud es probablemente la variable biométrica más natural para explicar el peso de un ejemplar. Para estudiarla, combinamos dos enfoques: una medida de asociación global y un modelo lineal simple sobre el logaritmo del peso.
3.2.1) Correlación global entre longitud y peso
cor_longitud_global_pearson <- cor(datos_ieo$Longitud_cm, datos_ieo$Peso_g, method = "pearson")
cor_longitud_global_spearman <- cor(datos_ieo$Longitud_cm, datos_ieo$Peso_g, method = "spearman")
cat("Correlación de Pearson (Longitud, Peso) =", round(cor_longitud_global_pearson, 4), "\n")
cat("Correlación de Spearman (Longitud, Peso) =", round(cor_longitud_global_spearman, 4), "\n")Las correlaciones globales son muy elevadas: 0.8656 para Pearson y 0.9951 para Spearman. Esto indica una asociación muy fuerte entre longitud y peso y sugiere que, cuanto mayor es el ejemplar, mayor tiende a ser también su peso individual.
3.2.2) Modelo lineal simple con longitud
modelo_longitud <- lm(log_Peso_g ~ Longitud_cm, data = datos_ieo)
summary(modelo_longitud)
El modelo lineal simple con longitud confirma esta idea de forma aún más clara. El ajuste sobre log(Peso_g)
alcanza un R² ajustado de 0.9069, lo que significa que más del 90% de la variabilidad del peso transformado
queda explicada únicamente por la longitud. Entre los factores analizados, este es, con diferencia, el resultado más fuerte.
3.3) Asociación entre edad y peso
La edad representa otro posible factor explicativo del peso, pero aquí aparece una limitación importante del dataset: solo disponemos de edad válida para la submuestra de jureles. Por ello, el análisis de la edad debe interpretarse como un estudio parcial y no como una comparación completa entre especies.
3.3.1) Correlación global entre edad y peso
cor_edad_global_pearson <- cor(datos_edad$Edad, datos_edad$Peso_g, method = "pearson")
cor_edad_global_spearman <- cor(datos_edad$Edad, datos_edad$Peso_g, method = "spearman")
cat("Correlación de Pearson (Edad, Peso) =", round(cor_edad_global_pearson, 4), "\n")
cat("Correlación de Spearman (Edad, Peso) =", round(cor_edad_global_spearman, 4), "\n")Incluso con esta limitación, la relación edad-peso en la submuestra disponible es muy intensa: la correlación de Pearson es 0.9847 y la de Spearman 0.9528. Es decir, dentro del jurel, los individuos de mayor edad tienden también a presentar mayor peso.
3.3.2) Modelo lineal simple con edad
datos_modelo_edad <- datos_ieo %>%
filter(!is.na(Edad), Edad >= 0)
modelo_edad <- lm(log_Peso_g ~ Edad, data = datos_modelo_edad)
summary(modelo_edad)El modelo lineal simple con edad alcanza un R² ajustado de 0.7726. Se trata de un valor alto, lo que sugiere que la edad también está fuertemente asociada al peso. Sin embargo, debemos ser prudentes: este resultado se obtiene sobre una submuestra más reducida y formada únicamente por jureles, por lo que no es directamente comparable con los modelos ajustados sobre la muestra completa.
3.4) Comparación preliminar de factores explicativos
Para responder de forma más directa a la pregunta central del trabajo, comparamos varios modelos sencillos construidos sobre
log(Peso_g). La idea aquí no es construir un modelo predictivo final, sino valorar cuál de los factores
considerados parece aportar mayor capacidad explicativa de manera aislada.
datos_modelo_base <- datos_ieo %>%
filter(!is.na(log_Peso_g), !is.na(Longitud_cm)) %>%
droplevels()
datos_modelo_edad <- datos_ieo %>%
filter(!is.na(log_Peso_g), !is.na(Longitud_cm), !is.na(Edad), Edad >= 0) %>%
droplevels()
modelo_especie <- lm(log_Peso_g ~ Especie, data = datos_modelo_base)
modelo_longitud_comparable <- lm(log_Peso_g ~ Longitud_cm, data = datos_modelo_base)
modelo_edad_comparable <- lm(log_Peso_g ~ Edad, data = datos_modelo_edad)
comparacion_modelos <- data.frame(
Modelo = c("Especie", "Longitud", "Edad"),
n = c(nrow(datos_modelo_base), nrow(datos_modelo_base), nrow(datos_modelo_edad)),
R2 = c(
summary(modelo_especie)$r.squared,
summary(modelo_longitud_comparable)$r.squared,
summary(modelo_edad_comparable)$r.squared
),
R2_ajustado = c(
summary(modelo_especie)$adj.r.squared,
summary(modelo_longitud_comparable)$adj.r.squared,
summary(modelo_edad_comparable)$adj.r.squared
),
AIC = c(
AIC(modelo_especie),
AIC(modelo_longitud_comparable),
AIC(modelo_edad_comparable)
)
)
comparacion_modelos <- comparacion_modelos %>%
mutate(
R2 = round(R2, 4),
R2_ajustado = round(R2_ajustado, 4),
AIC = round(AIC, 2)
)
print(comparacion_modelos)La comparación preliminar es muy reveladora. El modelo con especie alcanza un R² ajustado de 0.3488, el modelo con longitud llega a 0.9069 y el modelo con edad alcanza 0.7726. Esto sugiere que, entre los factores considerados, la longitud es la variable individual que mejor explica el peso. La edad también muestra una asociación elevada, pero sobre una submuestra más limitada, mientras que la especie, aun siendo relevante, queda claramente por detrás en capacidad explicativa aislada.
Además, el modelo combinado con especie, longitud y edad no pudo ajustarse de forma comparable porque en la submuestra con edad válida solo quedaba una especie. Este detalle no es menor: limita la posibilidad de construir un modelo único con todos los factores en igualdad de condiciones y obliga a interpretar los resultados de la edad con más cautela.
3.5) Qué nos deja la inferencia
En conjunto, la parte inferencial refuerza y matiza lo observado en el análisis exploratorio. La especie introduce diferencias estadísticamente significativas en el peso medio; la merluza es, en promedio, mucho más pesada que el jurel. Sin embargo, cuando tratamos de valorar qué variable parece explicar mejor el peso individual, la evidencia apunta de forma bastante clara hacia la longitud.
Dicho de otra forma: la especie importa, pero la longitud parece capturar una parte mucho más grande de la variabilidad del peso. La edad también muestra un patrón fuerte, aunque con una evidencia más limitada por la estructura de los datos disponibles. Con esto ya tenemos una base sólida para cerrar el estudio y extraer una conclusión general coherente con la pregunta de partida.
4) Conclusión general
Después de recorrer todo el flujo de trabajo —desde la extracción de los datos y su depuración hasta el análisis exploratorio y la parte inferencial— ya podemos responder con bastante claridad a la pregunta que guiaba el estudio: ¿qué factores parecen explicar mejor el peso individual de los ejemplares analizados?
En primer lugar, la especie sí desempeña un papel importante. Tanto los gráficos como los intervalos de confianza y el contraste t de Welch muestran que la merluza presenta un peso medio claramente superior al del jurel. La diferencia media estimada entre ambas especies fue de 428.18 gramos, y el intervalo de confianza al 95% para la diferencia de medias fue (366.15, 490.21), completamente positivo. Por tanto, existe evidencia estadísticamente significativa de que ambas especies difieren en su peso medio.
Sin embargo, cuando ampliamos el foco y dejamos de preguntar solo si una especie pesa más que otra, el resultado más
interesante del estudio aparece al analizar la longitud. Tanto visualmente como mediante correlaciones y
modelos lineales sencillos, la longitud muestra la asociación más fuerte con el peso individual. En particular, el modelo
lineal simple sobre log(Peso_g) con la longitud como predictor alcanza un R² ajustado de 0.9069,
claramente superior al obtenido por el modelo basado únicamente en la especie.
La edad también aparece como una variable potencialmente relevante. En la submuestra donde fue posible analizarla, la relación edad-peso fue intensa y el modelo lineal simple con edad alcanzó un R² ajustado de 0.7726. No obstante, este resultado debe interpretarse con cautela, ya que tras el filtrado solo quedaron edades válidas para el jurel. Esto impide comparar el efecto de la edad entre especies en igualdad de condiciones y limita el alcance de esta parte del análisis.
En conjunto, los resultados sugieren que el peso individual no depende de un único factor, sino que refleja una combinación de características biométricas y biológicas. Aun así, dentro de las variables consideradas en este estudio, la longitud parece ser el factor individual con mayor capacidad explicativa, seguida por la edad en la submuestra disponible, mientras que la especie sigue siendo relevante, aunque con una capacidad explicativa aislada claramente menor.
Más allá de la conclusión biológica concreta, el trabajo también deja una idea metodológica importante: con datos reales, la calidad y disponibilidad efectiva de las variables condicionan mucho más el análisis que el planteamiento inicial. Precisamente por eso, combinar limpieza de datos, análisis exploratorio, intervalos de confianza, contrastes de hipótesis y modelos lineales sencillos nos ha permitido construir una respuesta razonable, matizada y estadísticamente fundamentada a partir de una fuente oficial como DATRAS.
Descarga el Rmarkdown
Si quieres replicarlo exactamente igual y jugar con él, aquí tienes el rmarkdown completo listo para ejecutar.
Fuentes
- Instituto Español de Oceanografía (IEO-CSIC), organismo público de investigación responsable de la ejecución de campañas oceanográficas como DEMERSALES y de la recolección biológica en origen. ieo.csic.es
- ICES / CIEM (Consejo Internacional para la Exploración del Mar), organización intergubernamental que centraliza, coordina y publica datos marinos en el Atlántico Norte. ices.dk
- Base de datos DATRAS (Database of Trawl Surveys), repositorio del ICES del que se extrajeron los registros biológicos del estudio. datras.ices.dk
-
Paquete
icesDatraspara R, utilizado para consultar de forma programática la API web de DATRAS. cran.r-project.org